ANDA制剂研发中预BE试验的实施探讨(雷继锋,郭汉江,李冠谕等老师倾情分享)
[导读]
无论是制剂国际化的遍地兴起,还是一致性评价的全体参与,都将中国的制剂研发推向一个前所未有的热度。作为仿制药研发的核心模块,生物等效性试验成为时下医药产业界最热门的话题。要不要进行预BE, 业界对此有不同的看法,预BE如何实施,也存在诸多疑惑。
2016年9月,CninMed (医药制剂国际化) 邀请ADNA制剂专家葛季声、杨东、汤丽娟及褚襄萍等博士,以及SAS公司高级顾问陈家松先生对ANDA制剂研发CMC模块的QbD与DOE进行了深入探讨,讨论文字稿 “ANDA制剂研发中QbD与DOE的运用” 为ADNA制剂研发提供了很好的实际操作指南。
2016年10月,CninMed继续邀请业内专家雷继锋先生,郭汉江先生,李冠谕先生,以及梁舜华先生和大家就ANDA制剂研发中的关键模块-BE试验,结合更多实际案例进行了“ANDA制剂研发中预BE试验的实施探讨”。
[主要参与嘉宾]
雷继锋先生:
上海安必生制药创办人及CEO。曾先后任职于西安杨森、赛诺菲和葛兰素史克核心管理岗位。2007年创办上海安必生制药,帮助中国药企申报俞20多个ANDA,帮助3家中国企业和1家日本企业通过FDA的GMP现场检查。2015年底至2016年初,上海安必生获得孟鲁司特钠片与孟鲁司特钠咀嚼片两项ANDA批准。雷老师具有制药全领域的丰富实际经验,他率先切入制剂国际化领域,更是探路中国MAH制度的先行者。
郭汉江先生:
台湾资深制剂产品开发前辈。郭老师1984年开始进入台湾制药行业,具有俞30年的制药职业生涯。退休后2009年至2011年在东南亚制药企业担任顾问。2011年至今服务于现台湾公司。郭老师擅长口服缓控释制剂如骨架缓释片,膜控缓释片,渗透泵片,缓释胶囊等产品开发。累计提交TFDA台湾药品申报200件以上,大陆CFDA申报10件以上,向 FDA申请ANDA超过20件以上,其中不乏挑战专利申报,另有创新产品505(b)2超过5件以上。
李冠谕先生:
台湾鸿谕药品生技创办人。药代动力学方面的资深专家,精通于BE实验。李老师1997年开始进入台湾CRO产业。2003年参与中国药代研讨会后,开始长期和大陆制药业界交流合作。2006年在台湾创立临床试验CRO公司鸿谕药品生技,辅导过两个临床基地的建立,并具有建立GLP认证药代分析实验室的经验。同时提供大陆与东南亚制药企业咨询及BE服务。李老师经验颇丰,专长于药代动力学之研究,生物等效性试验试验方案的设计和执行,生物检体的分析方法学开发,验证试验,运用生物资讯 (Bioinformatics) 模拟于新药开发。累计执行过超过400件Pilot BE试验及200件以上Pivotal BE试验。
[其他参与人员]
梁舜华先生:印度CRO公司中国业务负责人,梁先生及其公司主要为针 FDA, EMA, WHO的产品申报提供BE试验服务。
葛季声博士: 20多年美国知名药企制剂研发经验。美国V&M Pharmtech威和麦公司合伙人。曾担任亚宝药业北京研究院制剂研发总监及研究院副院长,负责亚宝药业仿制药国际化项目。
褚襄萍博士:上海海翔制剂研发总监。具有多家知名制剂国际化企业职业经历,具有丰富的制剂产品开放经验。
小望-望开鹏:制剂国际化制剂研发新兵。业余着力于医药制剂国际化跟踪与传播。“制剂国际化”系列讨论参与人。(微信加281660433参与更多交流)
[讨论提纲]
望开鹏:现在制剂国际化和一致性评价同时都很火热,作为制剂产品开发的核心模块,生物等效性试验空前热门。关于BE临床机构难找,BE报价水涨船高,预BE试验通过率低等各种传闻都反映了国内生物等效性试验执行上的一些情况。现在我们放眼大陆之外,基于ANDA制剂研发,来探讨生物等效性实验的相关问题。
讨论主题:ANDA制剂研发中预BE试验的实施探讨。
1, 预BE试验有没有必要及理由,预BE的指导意义有多少。
2, 从产品本身,也从预算考虑。什么情况下预BE试验很有必要。
3, 时间、费用、合规。印度、美国及加拿大预BE试验一般要求及考量。
4, 预BE试验的设计考虑和结果数据的判断分析,及申报时数据提交的法规要求。
由于法规的差异,我们的讨论主要基于ANDA研发中的预BE试验。由于时间紧凑,部分问题不易展开,后续可继续交流。以下是讨论内容的精简编辑。
1 [预BE实验的意义及必要性]
雷继峰:
我尝试回答第一个问题,预BE试验有没有必要,它的指导意义是什么。大家都知道,仿制药是仿制原研药,但是我们做的仿制药,由于某种原因,比如挑战或规避处方工艺专利;或者没有某种原研的辅料,需要更换其他的辅料替代;或者为了节省成本,采用的工艺不一样;还有各种原因,导致仿制药的处方工艺和原研药不一样,尤其是缓控释产品,我们需要做个预BE。
但是不是说所有的药都需要做预BE,比方说一个速释的产品,同时是BCS I类或III类药物,预BE试验就没有太大的必要。
对于缓控释制剂,尤其是挑战专利的缓控释制剂,个人建议还是应该做预BE试验。
李冠諭:
補充一些,部分的品种预BE是没有必要的,可以用溶出曲线来判断,直接进行主BE。
在FDA的规范中提到:申请者在进行一个完整的BA/BE试验之前,可以先进行小样本数的预试验进行初步研究。试验研究可用于验证分析方法,评估PK变异性,确定样本数的规模大小以获得足够的统计效力,优化检体采集时间的间隔,以及确定给药周期之间有足够的清除期时间。
具体来说,通常预BE的指导意义是为了确认以下几点:
1-1 药代动力学的采血点设计:
可以优化样品收集时间间隔,清除期(洗脱期)的时间是否足够;
药代动力学曲线能够完整表现吸收、分布、代谢、排除为最佳;
1-2 生物样品的分析方法学是否恰当
可以验证分析方法的合适性。例如:分析线性范围是否合适,代谢产物的分析方法是否合适;
1-3 药品的变异性:
以预BE的药代参数结果进行主BE的人数模拟;
确定样本数大小以获得足够的成功机率;
预BE除了确认制剂处方无太大差异外,其结果以统计学来做为主BE人数的推估是相当准确的。
雷继峰:
正如李老师所讲的,预实验作用和目的,一个看是处方好不好,第二个是看一下,血药浓度的采血点合理不合理?第三看一下它的变异的情况。
我要特别特别说明的是,预实验因为例数比较少,从统计学的角度上,不太可能达到90%的置信区间在80%-125%之间,主要是从统计学上看平均值在不在合理范围之内。
梁舜华:
我赞同雷总的说法。作为仿制药来说,做BE是一个最终的标准,因为在体外无法完全模拟体内的吸收、分布等,做一个预实验是最直观的研究的结果的一个过程。
预实验是一个手段,也可以算是一个研发的一个阶段,而是否使用这个工具,就看具体制剂研发人员对相应产品的一个把握了。而且做预实验的时候,对产品有一个比较深入的理解,能够知道哪些因素对产品的PK行为有一定的了解。这样拿到数据以后,就能够更好的了解数据,预实验也就更加有指导意义了。
葛季声:
同意雷老师说法,对复杂剂型和BCS 2类几乎必做预BE, 当然也可直接做BE,但失败可能性很大。预BE在美国仿制药公司是常态,以后在国内也应成“新常态”。
溶出曲线是做制剂的一个很有用的工具,尤其是BCS 1、3类。但是对缓控释剂型,特别是当制剂配方组分需要改变时,会出现溶出曲线对不上而BE能通过的可能。科学原因:不同辅料对API吸收的影响是不同的。这也是为何需要做预BE的道理。
郭汉江:
同意雷總的意見。嚴格上來說,預BE是風險前的評估,如果沒有預BE則必須評估BCS Class。執行預BE是風險評估的一個重要機制,要不要執行預BE取決於處方研究前對於API性狀的了解程度。
有關BE預試驗問題,还主要會受限於廠家的資金與風險管理以及時間的問題!到目前為止應該沒有幾家公司可以直接去冒險執行BE正式試驗吧!
目前FDA對於Pivotal BE在查廠時,如果沒有預試驗數據都會嚴格檢查。
[补充案例] 某口腔崩解片,原研为冷凍乾燥法,在生产成本上偏高。修飾成一般口崩錠在製程上沒問題,但在溶離率上會相差很大,这时需要執行BE預試驗。
贺星华:
要与不要都有一定道理,做不做还是郭老师说的,财力,时间,风险,看你对这个产品的认知程度和能接受的预期结果来决定是否做预BE。速释口服固体制剂我们都直接做正式BE试验,没有预试验。
梁舜华:
其实正确的说法,预BE其实也是一个BE试验。从BE试验上面讲,预实验只是受试者数量比较少而已,正确的操作应该是跟一个正式试验应该是一致的,这样才能减少误差。
葛季声:
同意,预BE只是为省费用人数少而已,好的预BE应能指导正式BE,也有预BE通过但正式BE失败的例子。
褚襄萍:
对于处方和工艺都不变的BCS2类,做BE的CRO又正好已经有了经验,预BE的意义?
葛季声:
个人觉着这种情况可直接做正式BE。
雷继峰:
BCS 2类速释产品,如果没有专利问题,处方工艺和原研一致,同时体外也做了很多工作。这个时候可以做预BE, 也可以不做预BE。
梁舜华:
是否通过不是预实验的一个首要目的。个人觉得预实验的首要目的一个是对配方在人体中的进一步确定,一个正式试验上面的预演。这样可以确定采血点的设置是否正确、是否有可能出现一些不能预测的情况等。如果预算允许,个人还是觉得进行一个预实验是一个比较明智的选择。
汤丽娟:
我个人认为,如果预BE对于CRO来说是BE实验方法学确认的一个手段,每个BE实验前都需要。如果作为对制剂是否能够通过BE实验的预测手段,则很多情况不需要,而且要慎重。如果每个配方都用预BE来测试一下,是对产品及BE实验本身沒有理解的表现。
2 [Food Effect品种预BE试验的执行策略]
王峰:
请问各位,预BE时如何选择Fast or Fed State给药? 对于明确有Food Effects的品种,预BE时就两种给药都得做呢?
张玉:
Food Effect的产品,个人建议有条件还是做2个。因为任何一个通过,不能保证另一个也通过。这个其实是信心与经济承受能力,以及开发时间限制的问题了。先做一个也行,数据好也要再做空腹又要耗时且有不过风险。万一遇上,再调整处方时Fed还是要做。所以个人建议做2个。
郭汉江:
BCS Class II产品,常見問題是空腹通過,食物下沒有通過,或是相反。預BE執行方式取決於合約內容,可以先執行空腹,通過後就繼續執行食物,沒通過就停止。Oxybutynin,Palperidone,Quetiapine,都有Food effect。
张玉:
同意,合约是这么定的。
梁舜华:
这要看按照什么的要求来做的。可以按照做相应的要求,在报价合同的时候就应该做相应的规定。
建议先做Fed的。一般餐后的影响会大一些。餐后通过了,餐前通过率相对比较高。一般Fed通过了,大多数人也是把Fast也做一下。但是也有一些公司直接Fed通过了,就不做了。但是如果Fed出来的结果不好,就不会再进行空腹了。不过预实验没有通过的说法。只有是数据好跟不好。
代孔恩:
是,预BE非统计意义,但可明晰剂型药代参数在变异范围内的波动性。
李冠諭:
@王峰郭老师的建议您可以参考,可以先執行空腹,通過後就繼續執行食物,沒通過就停止。
葛季声:
印度做BE food effect,对食品的要求同FDA有无差别?
李冠諭:
BE food effect, CRO 都可以被要求参照FDA的标准。
望开鹏:
印度人和欧美人的饮食结构有巨大差异。做餐后实验,虽然按FDA的标准餐,但印度受试者的肠胃习惯和欧美有差异。有没有存在人种和饮食习惯影响BE结果差异的情况。
李冠谕:
饮食,人种都会有影响,但不影响BE的结果。食物影响除非是辅料增加吸收的影响,相同饮食不会有差异,有时候为了规避专利,使用不同辅料时可能会影响。
3 [多处方开展预BE试验的考虑]
王峰:
在哪些情况下,为加速开发进程,可考虑执行2 个或以上的自制制剂T1、T2 vs R的预BE?
李冠諭:
两制剂间有差异时,选择2个可控工艺的制剂,以RTT设计做处方筛选,进行3向交叉。前提要能够可控的处方调整。
梁舜华:
那要对某个因素有一个比较大的了解。有客户对一个制剂,认为其中的粒径因素是一个最主要的因素。他们就设计了两个不同粒径的配方来做预实验。
代孔恩:
TT差异如何界定,这样失败了,不是比RT预BE失败更难找原因。
张玉:
TT建议只改一个变量,否则多变量结果出来的差异性不知道是哪个引起的。
4 [预BE在挑战专利及建立IVIVC的运用]
望开鹏:
郭老师具有一些挑战专利的经验,也是否会设计不同处方同时预BE。
郭汉江:
會,至少三個處方。執行第一次預BE之後就必須建立IVIVC。必須有IVIVC才能通過。執行預BE最多二次就要完成通過。缓控释品种也是最多2次。但要取決有幾個處方要做,主要都是執行三交叉。
我提一個挑戰專利PIV的經驗。Quetiapine furamate er tablet喹硫平缓释片。這產品大家應該很熟悉。我在當地執行預BE, 一次就通過,但也有公司做3次也沒有通過。
李冠諭:
喹硫平会因为副作用影响吸收的状态,失败的多。
代孔恩:
@郭老师,如果执行RTT三交叉,在TT之间如何分辨出差异主要思考点是?
郭汉江:
取決於處方的差異。預BE 是否可以通過取決於對API溶解度的了解。如果處方開發前對於產品背景的了解後,就可評估處方是否能穩定與可通過預BE的結果了。溶解度的溶解與體內胃腸道吸收有直接關係。我主要專業在處方研究前做詳細資料整理與分析,就可確定通過率了。寧可錢花在處方研究,也不要冒險去執行預BE。
褚襄萍:
设计三个处方做预BE,建立IVIVC,再通过IVIVC来指导处方调整。
代孔恩:
也就是还是依靠预BE确定一个IVIVC来定量体外释放和体内吸收关联性,然后做一个体外释放跨距夹逼出释放范围。
王峰:
但有时建立IVIVC的代价比2轮预BE还高啊。
褚襄萍:
但是据统计,被FDA接受的IVIVC数据并不多。(补充说明:从1996年到2014年,FDA总共收到14个IVIVC申请,用在缓释制剂处方/生产地点/溶出条件变更,11个数据被拒绝,3个数据被接受,3个之中只有1个达到了目的。数据来源《杨劲,如何提高人体生物等效性实验的通过率》)。
郭汉江:
是的。IVIVC是廠家自己建立的,FDA不會要求。
5 [印度对预BE试验的要求]
望开鹏:
我们开始谈第3个问题。目前做预BE试验,试验地点比较多的是印度,那么印度对预BE实验有一些什么基本的要求?
雷继峰:
我来谈谈对第三个问题的看法! 其实无论是预实验,还是正式BE试验,都是用人体来做,所以各国都有伦理方面的要求,同时监管机构还有批准的要求。比方说,印度对于已上市的药品要多少年以后,和刚刚上市或者没在印度上市的药还是有不同的要求,对于速释和缓控释的产品也不同。
就政府批准来讲,他有一个实验样品进口注册叫做 T-license。还有一个批准临床实验,根据药品是速释还是缓控释,这个药品在印度上市了多久,要不要得到政府的批准,方案一旦得到批准以后,这个预实验就可以在印度CRO来做。
郭汉江:
目前印度要求執行預BE樣品必須在GMP廠房生產,也要有一個月加速數據。在印度執行預BE同樣要進行當地DCGI申请IRB審核通過,原則上只要求一個月加速。
梁舜华:
印度现在的要求还是比较简单的,一般分成需要NOC(相当于备案)和不需要NOC的产品。一般受试者通过的方案过了伦理以后,然后到DCGI(相当于CFDA)备案后,就可以开展了。
我准备了一个流程图,大家可以参考。
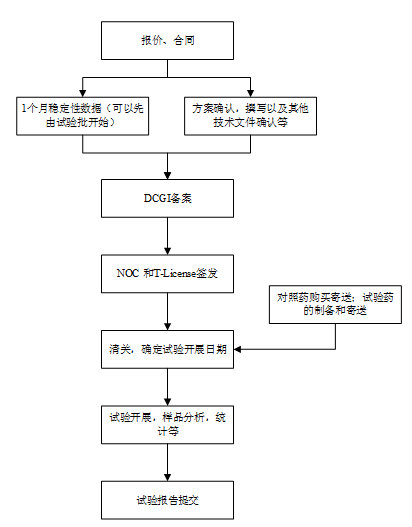
其中:
DCGI为印度的药监机构,其功能相当于中国的CFDA;
NOC为 Notice of Compliance,相当于国内的临床研究批件;
Test (Import) License Application (T-License) 相当于一次性进口批件。
以上两者为备案成功后同时签发。
梁舜华:
对于需要NOC的产品,需要至少一个月的稳定性数据才能申报。其中稳定性数据包括一般稳定性(Real Time Stability)和加速试验数据。其实根据印度对稳定性数据的指导原则,开展试验的时候,还需要3个月的数据。申报的时候,可以先交一个月的数据。
6 [美国和加拿大对预BE的要求]
望开鹏:
美国和加拿大的要求相对于印度有什么差异。比如稳定性数据方面,GMP要求方面。
葛季声:
美国这边做预BE同正式BE 一样有GMP、GCP要求,可以用一个月加速稳定性数据。
望开鹏:
一般做预BE实验的样品批量以及制备环境有些什么样的基本要求。在实验室制备样品,或者手工包装的样品是否也可以进行预BE试验。
雷继峰:
这个预实验的样品也是用于人体,所以应该没有污染和交叉污染。但是CRO和临床机构并没有要求你必须在GMP的工厂生产。也就是说可以在实验室做预BE实验的样品,但做之前,应该充分的确认这个产品有没有交叉污染的可能。把设备和环境都要进行清洁确认。还有刚才讲到的稳定性一个要求。另外,可能有些CRO或临床机构要求有一个GMP符合申明。
张玉:
一般预BE在中试或以上规模做比较有意义,所以实验室规模做的不多。
葛季声:
实验室如果符合cGMP,也应该也行。手工包装是可行的,但需要有生产和包装批文件、符合QC检验、QA放行要求。
7 [BE试验地点的选择]
葛季声:
@李冠谕现在亚洲特别是大陆这边做BE被FDA接受吗?。
陆慧:
这个问题我咨询过FDA的老师,对Site没有特殊要求,但人种如果有差异,方案设计需要一定考虑。
李冠谕:
是的,可以的,只要符合FDA的规范。
郭汉江:
我们目前预BE有些在上海做,也有印度做,主BE试验在印度做。
8 [预BE试验设计的一般考虑]
望开鹏:
由于时间关系,我们简单谈第四个问题,预BE试验的设计考虑和结果数据的判断分析,及申报时数据提交的法规要求。
李冠諭:
预BE试验的设计考虑,个人认为最重要的是以下3点:
1. 确认两制剂间无差异,双向交叉设计;
2. 两制剂间有差异时,选择2个可控工艺的制剂,以RTT设计做处方筛选,进行3向交叉;
3. 要有足够做统计仿真的样本数,至少8人建議12人。
梁舜华:
预试验一般做12例,如果变异系数大的,可以做18例,我们这边对于那些变异系数特别大的那些,甚至预试验做到24例的。
9 [预BE试验结果的判断分析]
李冠諭:
预BE结果资料的判断分析,需确认以下几点:
1 溶出曲线与药代血中浓度曲线的比对;
2 临床不良反应;
3 计划书偏离的纪录;
4 分析方法线性范围合适性;
5 药代参数与文献的差异比较;
6 离群值的确认;
7 进行主BE统计人数模拟。
王峰:
请问各位老师,预BE结果拿到后,预BE试验结果可否甄别主药在GI中存在特定吸收部位?如何从预BE试验的结果来反推最有区分力的体外溶出条件或组合条件?
梁舜华:
我认为还是一般情况下是比较困难的。或者运用一些比较特殊的手段,如放射性标记的药物,然后全程监控。比较困难。预实验能够一定程度体现这个配方和工艺的所造成的pK行为。而从pK行为反推配方,需要药剂方面的研究人员讨论解决了。
李冠諭:
可以用预实验的药代血中浓度曲线与制剂溶出曲线比对。
张玉:
溶出曲线与药代血中浓度的对比这块,是否可以稍微深入的谈一下?
李冠諭:
一般来说,胃排空2小时,会以 pH 1.2为主,Tmax 在1.5-2小时的品种可以明显与溶出的结果做比对。
郭老师:
目前對於IVIVC的課題是一定要建立的。首先必須確認是否有IVIVC ? 同時要確認藥品間的偏差大小。有些產品執行預BE,會像是翹翹板原理,一個通過一個沒通過。
郭老师:
這個翹翹板問題是目前存在的共同問題,必須從處方與工藝進行調整與修飾。個人建議從開發產品資料整理分析與處方試製品質控制以及溶離率不同方法分析需建立嚴謹才能降低後續的問題。
雷继峰:
@张玉 在做预BE之前呢,通常都会做4条溶出曲线,在相关介质的溶出。基本上差不多了才去做预实验,但预实验的结果出来可能和想象的不一样,可能高,低,有时候还差得很多。这时候就回去探索,看原来考虑的哪些条件?重新探索新的溶出条件,看看哪一个溶出条件和预实验对的上。对应上以后,再去按这个处方,在新的处方正式BE的时候通过了,那证明预实验PK有一定的相关性,这个最多证明有IVIVR。但IVIVC是要通过实验数据反卷积的方法得到体内的溶出曲线和你刚才的溶出曲线比,这样的才叫IVIVC。 FDA的IVIVC也有不同级别的。
褚襄萍:
雷总提到预BE对于处方的意义在于,提供可能的IVIVC或者IVIVR信息,又回到Bio相关的溶出方法对于处方开发的重要性了。那么可能BCS 2类做预BE才有这方面的意义。BCS 1类和3类就没有这方面的意义了。
10 [预BE试验数据申报的法规要求]
望开鹏:
预实验数据,这个数据结果是不是也需要申报时提交到FDA。
张玉:
ANDA申报材料中有预BE研究情况部分。
梁舜华:
现在FDA的要求,做过预试验的,都会要求提交。
李冠谕:
需要提交,FDA在修订BE数据规则中清楚的规定,每次进行研究都需要提交完整或总结报告。
[结束语]
望开鹏:
各位同仁,由于时间关系,我们今天的讨论就暂时告一段落。非常感谢雷继峰老师,郭汉江老师,李冠谕老师,以及梁舜华先生的分享和参与,也感谢褚襄萍博士和葛季声博士的热情支持。今天的讨论内容非常精彩,相信会给大家在预BE试验方面提供了有用的参考。
[BE试验补充分享]
李冠谕:
BE要两制剂在同一个个体内比较才能减低实验上的变数,除非有伦理或安全上考量,才需要特殊族群。简单来说,BE就是自己跟自己比。一般BE实验都设计双向交叉,就是让个体间差异变小。
我举个案例给大家参考,阿立哌唑T1/2约75小时,清洗期约21天。一般半衰期长的品种,会以平行试验设计来执行预BE,但因为P450酶CYP3A4或CYP2D6 EM PM的基因型差异会造成吸收的差异。因为预BE主要是确认两制剂在体内的变异系数,所以要执行交叉设计才能取得体内差异系数(Intra CV),平行试验设计预测结果所获得的变异性数据只是个体间的差异系数(Inter CV),无法确定制剂处方差异还是基因型差异造成的吸收差异。
再举一个实例,恩替卡韦(Entecavir)预BE执行交叉试验Intra CV 23%,预估交叉24例有85%的成功机率。但在委托印度CRO时,试验设计被改成平行48例,结果失败了。后来相同的处方,执行交叉设计,BE试验成功了。
贺晴:
@李冠谕确实BE做平行设计要比做交叉设计风险大,即使增加例数,因为个体间的误差在平行实验中会有影响。
李冠谕:
受试者的控制尽量去要求,从保护受试者的考量上,有一些变数是无法得知的,只能从统计学的角度去预防离群值的发生,降低影响BE结果。
男女不等性这部分,我再以经验分享给大家。美服培酮,在某些国家执行试验会有伦理考量,后来以男性做受试者给药,其结果BE通过。
西他那非,因为副作用,体内变异性从4年前的数据35%到现在的55%。尝试与伦理专家沟通,改全部由女性受试者入组,没获得同意。
李旭东:
我听说日本不考虑人种差异,因为他们做了几十年全科室的药物跟踪,大数据分析差异很小。
李冠谕:
就人种差异造成的部分,可以从药代参数看到结果,这些数据是为了临床使用上的参考。
日本以往要求BE,一定要日本人执行。近几年才听说要修正,还没见到具体规范。
人种上就药代,疗效,一般不会有太大差异。临床运用上是持续多剂量给予,就要考虑安全性。印度与美加都是广义的高加索人种,印度是雅利安人是高加索的一个分支。阿拉伯人也是一个分支。其实只要做Y染色体单倍群的检测就可以辨别人种。
[重要观点补充]
雷继锋:
关于我国接受海外临床机构BE试验数据一事,容我提供一些科学和法规依据,供大家讨论参考。
第一,仿制药生物等效试验(BE)是比较两个处方在同一个人体(受试者)上的体内表现。BE试验结果与受试者人种种族无关,只与处方工艺相关,欧美和WHO专家对此认识完全一致。我国医药科学界,医药监管界,医药产业界在此点上不应有争论和歧义。BE试验结果与人种、年龄、体重、性别等无关。新药的PK结果与种族体重年龄性别等有关系。
第二,所有BE试验(药代,药效,体外和临床终点等)均应遵循GCP和GLP及数据可靠性(完整性)的相关要求。我国CFDA接受的国内外BE数据,均按此要求来判断合适否。
第三,以药代为终点判断的BE试验研究指南,我国CDE己在今年五月正式发布,该指南内容与美国FDA2013年更新的以药代为判断依据的BE指南内容完全一致(全面翻译整理的),说明我国BE标准己与国际BE标准接轨。
第四,具体产品的BE指南,美国FDA己公布近1500个产品的BE要求并及时调整更新,各国产业界和监管界普遍认为这些BE技术要求具有科学性,我国CDE也陆续翻译并参照采纳。
第五,欧美和WHO,我国台湾地区均接受世界任何国家符合GCP和GLP的BE数据。我国CFDA也应仿效,审核查验中心(FDl)派员检查海外的BE数据,这样做即体现大国风度和我国的GCP和GLP要求与世界同步,也可学习国外GCP和GLP的实践做法,提高我国对GCP和GLP的检查水平,促进我国临床试验研究的提高与国际接轨
[参考文献]
[1] Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted under an Abbreviated New Drug Application (ANDA). FDA. Dec. 2013.
[2] Guidance for Industry: Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs- General Considerations. FDA. March 2014.
[3] Guidelines for Bioavailability & Bioequivalence Studies. CDSO, India. March 2005.
[4] Guidance Document: Conduct and Analysis of Comparative Bioavailability Studies. Health Canada. 2012.
[相关书籍]
生物药剂学在药物研发中的运用. 宁保明,杨永健主译.
转载请注明:医药制剂国际化--Hi-Drug药聚旗下网站 » ANDA制剂研发中预BE试验的实施探讨(雷继锋,郭汉江,李冠谕等老师倾情分享)